
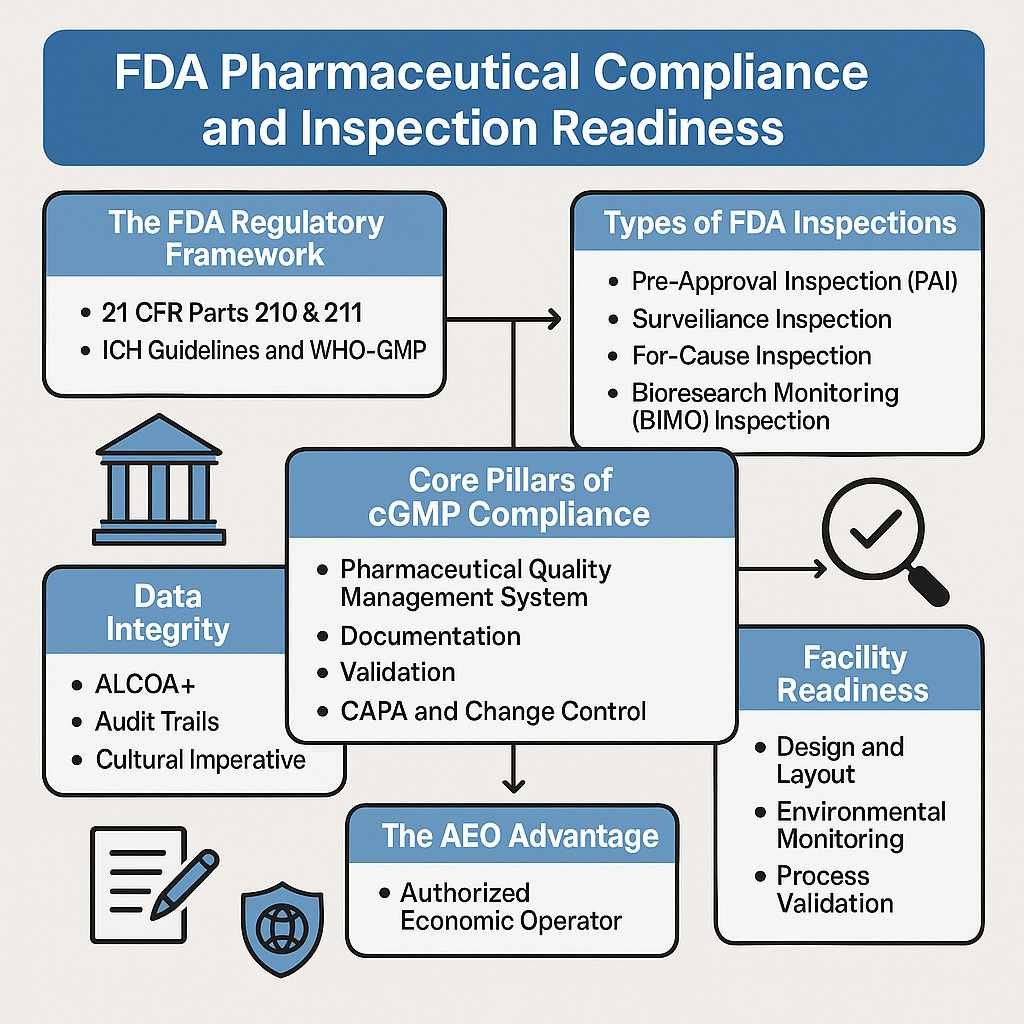

Entering the United States pharmaceutical market is not simply an expansion decision—it is a regulatory commitment governed by the U.S. Food and Drug Administration (US FDA). Every drug product marketed in the United States must comply with strict standards related to safety, efficacy, quality, labeling, and manufacturing practices.
Because the US FDA regulatory framework is highly technical, continuously evolving, and enforcement-driven, pharmaceutical companies increasingly rely on a US FDA Drug Registration Consultant to ensure compliant and timely market entry.
US FDA Drug Registration: A Regulatory Control System, Not a Formality
US FDA drug registration is not a one-time administrative task. It is a structured regulatory control system that governs how drugs are developed, manufactured, reviewed, approved, distributed, and monitored throughout their commercial life.
Every registered drug remains under continuous US FDA oversight, covering:
- Manufacturing quality systems
- Data integrity and documentation
- Product safety monitoring
- Labeling and promotional compliance
- Inspection readiness and enforcement response
A US FDA Drug Registration Consultant helps pharmaceutical companies align their internal systems with these regulatory control mechanisms from the very beginning.
Why US FDA Regulations Demand Expert Consultancy
US FDA regulations are interpretation-sensitive. Two companies manufacturing identical products can receive very different regulatory outcomes based on how data is presented, validated, and defended.
A US FDA Drug Registration Consultant provides:
- Regulatory interpretation based on current enforcement trends
- Structured submission strategies aligned with US FDA reviewer expectations
- Risk-based compliance planning
- Controlled communication with US FDA authorities
XPRO America, a US FDA Consultancy, operates as a regulatory extension of pharmaceutical organizations, ensuring alignment with US FDA expectations at every stage.
Regulatory Pathways Governed by the US FDA


Selecting the correct US FDA regulatory pathway determines submission complexity, cost, and approval timelines.
New Drug Application (NDA)
Applies to drugs with new active ingredients, new indications, or novel delivery systems. Requires comprehensive clinical and non-clinical evidence.
Abbreviated New Drug Application (ANDA)
Used for generic drugs demonstrating bioequivalence with an approved reference product, with emphasis on CMC robustness and study design.
Investigational New Drug (IND)
Mandatory before initiating clinical investigations intended to support future US FDA approval.
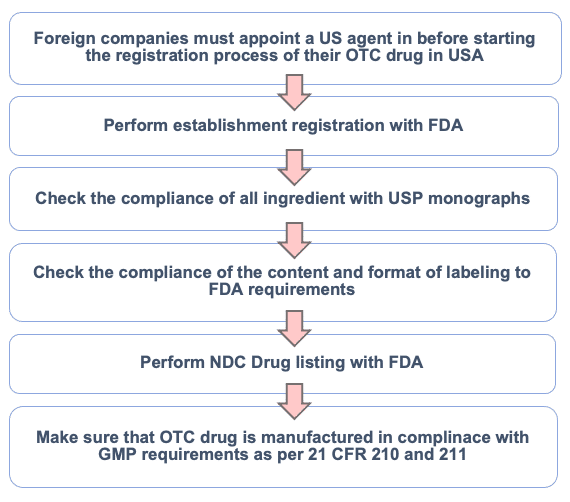
Over-the-Counter (OTC) Drug Submissions
Applies to non-prescription drugs, either under US FDA OTC monographs or via NDA depending on formulation and claims.
A US FDA Drug Registration Consultant ensures that regulatory pathways are selected strategically, not reactively.
Controlled Phases of the US FDA Drug Registration Process


Phase 1: Regulatory Intelligence & Gap Assessment
A detailed evaluation of product classification, formulation, manufacturing controls, and documentation readiness against US FDA requirements.
Phase 2: US FDA Establishment Registration
All facilities involved in manufacturing, testing, labeling, or packaging must be registered with the US FDA and renewed annually.
Phase 3: Drug Product Listing
Each drug product is listed with the US FDA, linked to registered establishments and assigned product identifiers.
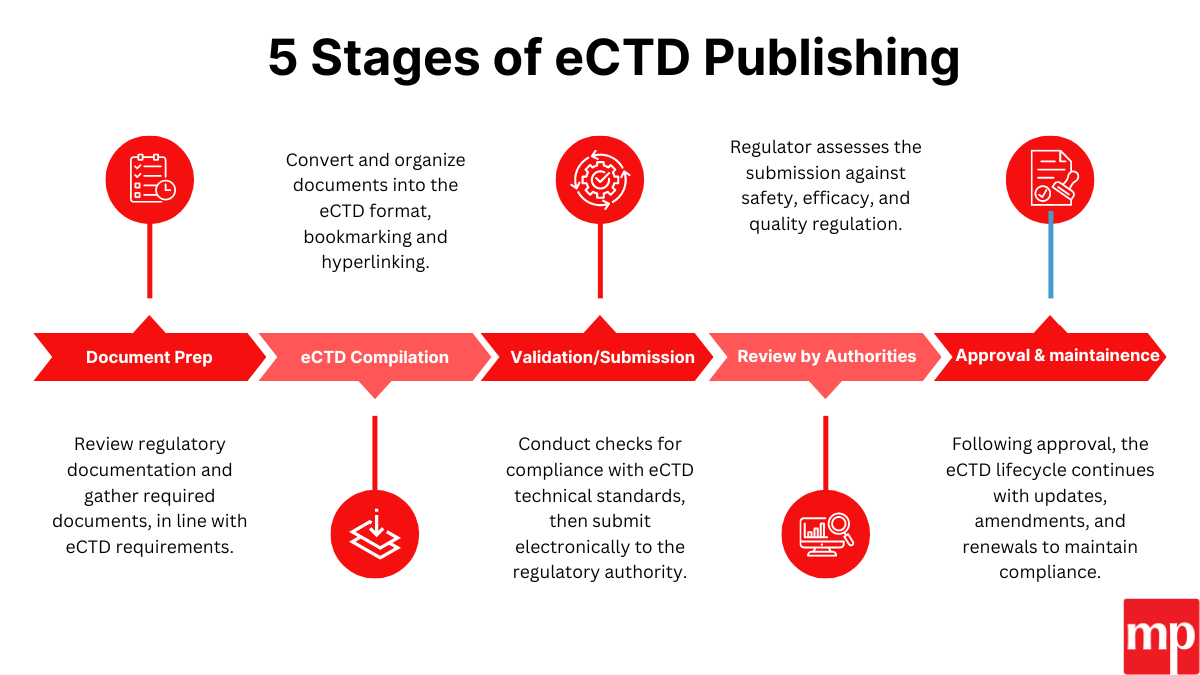
Phase 4: eCTD Dossier Development
Preparation of structured submissions in electronic Common Technical Document (eCTD) format, including:
- Chemistry, Manufacturing, and Controls (CMC)
- Stability and validation data
- Clinical or bioequivalence evidence
- US FDA-compliant labeling
Phase 5: US FDA Review & Technical Queries
US FDA reviewers assess submissions and issue information requests or deficiency letters that require precise, defensible responses.
Phase 6: US FDA Inspections & Audit Readiness
Facilities may undergo US FDA inspections to verify cGMP compliance, data integrity, and quality systems.
Phase 7: Approval & Post-Approval Lifecycle Compliance
Approval initiates continuous obligations such as pharmacovigilance, annual reporting, change control, and inspection readiness.
XPRO America, a US FDA Consultancy, manages this entire controlled lifecycle.
Regulatory Risks Without a US FDA Consultant

Organizations that approach US FDA registration without expert guidance often face:
- Technical rejections due to dossier inconsistencies
- Prolonged review cycles caused by unclear responses
- US FDA Form 483 observations
- Warning letters and import alerts
- Commercial delays and financial exposure
A US FDA Drug Registration Consultant focuses on risk prevention, not damage control.
XPRO America – Global US FDA Drug Registration Consultancy
XPRO America is a specialized US FDA Consultancy delivering global regulatory support for pharmaceutical companies seeking US market access.
US FDA Drug Registration Services:
- Regulatory strategy and submission planning
- NDA, ANDA, IND, and OTC drug submissions
- US FDA establishment registration and drug listing
- eCTD publishing and lifecycle maintenance
- US FDA query management and deficiency response
- US Agent services for foreign manufacturers
- Post-approval compliance and regulatory intelligence
XPRO America’s approach is compliance-first, audit-ready, and aligned with real-world US FDA enforcement expectations.
Strategic Advantages of a US FDA Drug Registration Consultant
Partnering with XPRO America delivers:
- Predictable regulatory timelines
- Reduced inspection and enforcement risk
- Stronger submission quality
- Clear accountability across the product lifecycle
- Long-term US FDA compliance sustainability
These advantages directly impact commercial success and brand credibility.
Who Requires US FDA Drug Registration Services?
US FDA drug registration consultancy is essential for:
- Pharmaceutical manufacturers
- Generic drug companies
- Contract development and manufacturing organizations (CDMOs)
- OTC drug marketers
- Emerging biotech firms
Any organization interacting with the US pharmaceutical market is subject to US FDA jurisdiction.
Continuous US FDA Compliance: A Business Imperative
US FDA approval does not eliminate regulatory exposure. Ongoing non-compliance can lead to:
- Product recalls
- Market withdrawal
- Regulatory sanctions
- Long-term reputational damage
A professional US FDA Drug Registration Consultant ensures that compliance remains continuous, proactive, and defensible.
Conclusion
US FDA drug registration is a high-stakes regulatory process governed by science, documentation integrity, and inspection readiness. Success requires more than submission—it requires regulatory strategy.
XPRO America, a US FDA Consultancy, provides globally aligned, technically precise, and enforcement-aware US FDA drug registration services designed for sustainable market access.
📧 For global US FDA drug registration consulting support, contact:
support@xproamerica.com
Leave a Reply